
When the FDA shows up at your generic drug manufacturing facility, it’s not a surprise visit-it’s a routine check. But that doesn’t mean you can wing it. The agency doesn’t just walk in and glance around. They’re looking for proof that every pill, capsule, or injection you make is safe, consistent, and meets the exact standards laid out in Current Good Manufacturing Practices (CGMP). And if you’re not ready, it can cost you time, money, and market access.
What Happens Before the FDA Even Arrives
Before any inspector steps through your door, your facility has already been flagged. The FDA doesn’t pick sites randomly. They use a risk-based model that looks at things like past inspection history, the type of drugs you make, complaints from patients or pharmacists, and even tips from insiders. Facilities making high-risk products-like injectables or drugs with narrow therapeutic windows-are inspected more often. If you’ve had a warning letter in the last two years, you’re on the short list.
Companies that want to get ahead are using the FDA’s PreCheck program, launched in 2024. This isn’t a shortcut-it’s a feedback loop. You submit detailed plans for your facility layout, quality systems, and manufacturing processes before you even start production. The FDA reviews it, gives you feedback, and tells you if you’re on track. It’s like a dress rehearsal for your inspection. Many manufacturers now treat this as mandatory, not optional.
The Six Systems the FDA Checks
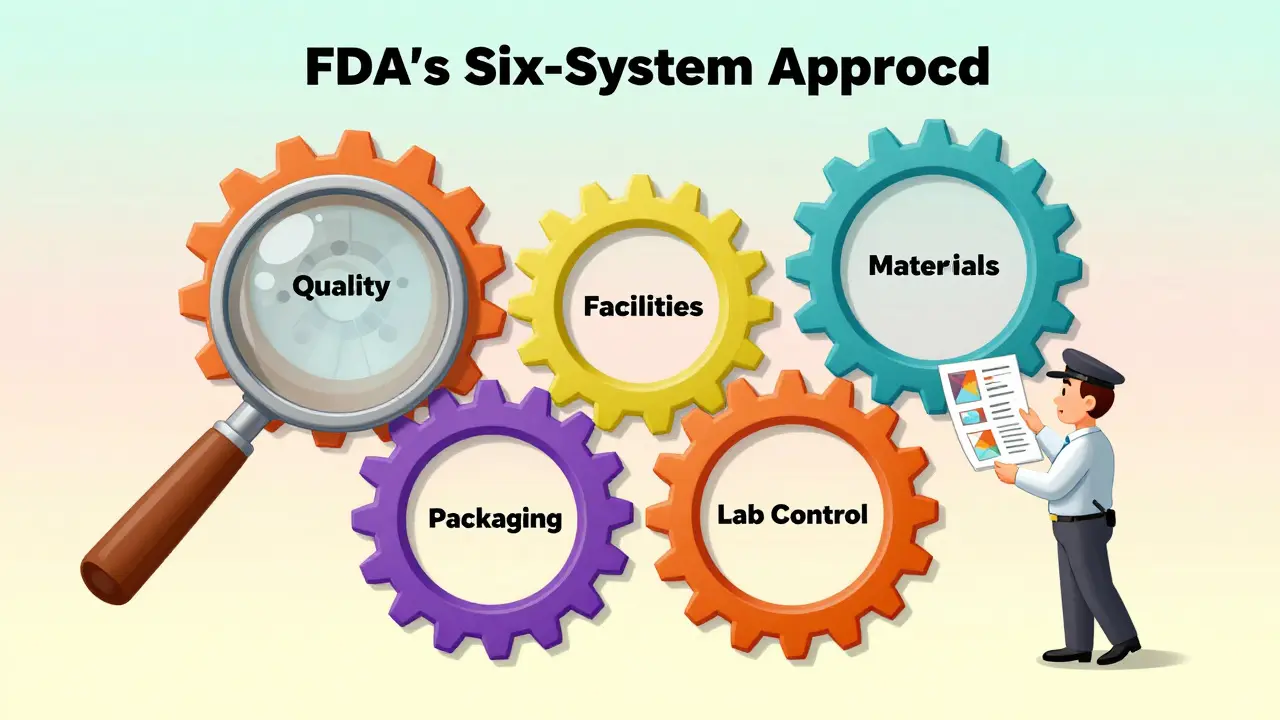
Every FDA inspection follows the same structure: the Six-System Approach. It’s not just about checking boxes. It’s about seeing how your entire quality system works together. The six systems are:
- Quality-Always inspected. This is the foundation. Do you have a dedicated quality unit that can stop production if something’s wrong? Are they empowered? Or are they just signing off on paperwork?
- Facilities & Equipment-Are your cleanrooms maintained? Are your equipment logs complete? Is calibration up to date? No one expects perfection, but they expect control.
- Materials-Where do your raw ingredients come from? Are your suppliers qualified? Do you test every batch of active pharmaceutical ingredient (API) before use?
- Production-Can you prove your process is validated? Do you have batch records that match what’s in your application? Are deviations documented and investigated?
- Packaging & Labeling-One wrong label can send a drug to the wrong patient. The FDA checks if your labeling matches your approved application and if you have controls to prevent mix-ups.
- Laboratory Control-This is where many facilities get tripped up. Are your analytical methods validated? Are stability studies being stored correctly? Is data being recorded in real time-or backfilled later?
The inspector won’t check all six systems in depth. But they will always check Quality, plus at least two others. And if something looks off in one area, they’ll dig into the others. It’s not about finding one big mistake-it’s about seeing if your system is broken from the inside out.
What They Look for During the Inspection
Inspectors don’t just ask for documents-they follow the trail. They’ll pick a batch number and trace it backward: from the finished product to the lab test, to the equipment log, to the raw material receipt, to the supplier’s certificate of analysis. If any link is missing, inconsistent, or altered, they’ll flag it.
They’ll look at your deviation reports. Not just how many you have, but how you handle them. Did you investigate the root cause? Did you change your process to prevent it from happening again? Or did you just close the report with “operator error”?
Data integrity is now the #1 focus. If your chromatography system shows a peak that was deleted, or if your balance logs have timestamps that don’t match your batch records, that’s a red flag. The FDA has trained inspectors specifically to spot manipulated data. They know how to read system logs, check audit trails, and spot patterns that suggest human interference.
They’ll also check your stability samples. Are they stored in the exact conditions you filed with your application? If your application says “2-8°C,” but the fridge is at 12°C, that’s a violation. No exceptions.
The FDA 483: What It Means and What to Do
If the inspector finds issues, they’ll give you Form FDA 483. This isn’t a notice of violation-it’s a list of observations. It’s your first warning. The observations are ranked by severity. The top one is usually the most serious.
Common 483 items include:
- 21 CFR 211.22(a): Lack of a qualified quality unit with authority
- 21 CFR 211.166: Inadequate testing of finished products
- 21 CFR 211.194: Missing or incomplete batch records
- 21 CFR 211.111: Failure to validate cleaning procedures
You have 15 business days to respond. Your response isn’t just an apology-it’s a plan. You need to show:
- What went wrong
- Why it happened
- How you fixed it
- How you’ll prevent it from happening again
Generic responses like “we’ve retrained staff” won’t cut it. The FDA wants evidence: updated SOPs, training records, revised validation protocols, corrected equipment logs. They’ll check your response against your actual practices during a follow-up.
What Happens After the Inspection
After the inspector leaves, they write the Establishment Inspection Report (EIR). This becomes part of your facility’s permanent record. If the findings are minor, your facility is marked as “acceptable.” That’s it-you’re cleared to ship.
If the issues are serious, the FDA may issue a warning letter. This is public. It’s posted online. It can block your product from approval. It can scare off customers. And if you don’t fix it, they can shut you down.
But there’s a path forward. In June 2025, the FDA finalized guidance for Post-Warning Letter Meetings (PWLMs). This lets you sit down with FDA officials, explain your corrective actions, and get direct feedback before you submit your formal response. It’s a chance to turn a failure into a learning opportunity.

How to Be Ready-Every Day
The biggest mistake facilities make is treating inspections like an event. They clean up the floor the week before. They print out extra SOPs. They tell staff to “be on their best behavior.”
That doesn’t work.
The FDA expects a state of control-not a state of readiness. Your facility should be inspection-ready every single day. That means:
- Quality isn’t a department-it’s everyone’s job
- SOPs are living documents, not dusty binders
- Deviations are investigated, not ignored
- Data is recorded at the time of the action, not later
- Training isn’t annual-it’s continuous
Run mock inspections every quarter. Bring in someone from another facility to play inspector. Ask them to pick a random batch and trace it. If they find gaps, fix them before the FDA does.
Facilities with mature quality cultures don’t just pass inspections-they impress them. The FDA knows the difference between a facility that’s trying to comply and one that’s committed to quality.
Bottom Line: It’s Not About Passing. It’s About Trust.
More than 90% of generic drug inspections find acceptable compliance. That’s not luck. It’s discipline.
The FDA isn’t trying to shut you down. They’re trying to make sure the medicine you’re making won’t hurt someone. If you’ve built a system where quality is baked into every step-from raw material to patient’s hand-you’re not just passing inspections. You’re earning trust.
And in the generic drug market, trust is the only thing that lasts longer than a patent.
Trevor Davis
January 13, 2026 AT 04:07Man, I’ve seen facilities go from zero to hero just by treating quality like a daily habit instead of a quarterly panic. The FDA doesn’t care if your floor sparkles on inspection day-they care if your data trails make sense. One guy I know started doing random batch tracebacks every Friday with his team. No warning letters since. Just consistency.
Lethabo Phalafala
January 14, 2026 AT 18:09Let me tell you something-this isn’t just about paperwork. This is about someone’s grandpa taking his heart medicine and not dying because the pill was made right. I’ve seen what happens when corners are cut. It’s not just regulatory-it’s moral. And if you’re not feeling that weight, you’re in the wrong industry.
Lance Nickie
January 15, 2026 AT 18:14cgmp? more like cgmpt-cant get my pants on today. why do we even do this? just print more pills and call it a day.
Milla Masliy
January 17, 2026 AT 14:18As someone who’s worked in pharma in both the US and India, I’ve seen how the same systems work differently across cultures. In the US, it’s all about documentation. In India, it’s about adaptability. The best facilities? They blend both-rigorous but human. The FDA respects that. Not perfection. Just integrity.
Damario Brown
January 19, 2026 AT 01:13you think data integrity is the #1 focus? lol. it’s the *only* focus. i’ve seen labs where people backfilled data from 3 weeks ago like it was a todo list. the FDA has AI tools now that detect timestamp anomalies better than your ex remembers your birthday. if your chromatogram has a ‘deleted peak’-you’re already on the list. no second chances. no ‘oops’.
Clay .Haeber
January 19, 2026 AT 07:49Oh wow, the FDA is soooo scary. They’re like the police of pills. What’s next? Are they gonna audit our coffee breaks next? ‘Sir, your espresso machine wasn’t calibrated to the 0.0001°C tolerance specified in Appendix G of the 2026 CGMP addendum.’ Get a life. Most of these inspections are theater. The real problem? Big pharma lobbying to keep generics expensive. The FDA’s just the puppet.
Priyanka Kumari
January 20, 2026 AT 13:06One thing I’ve learned working in India’s generic sector: quality isn’t a cost-it’s the only thing that lets you compete globally. We used to think ‘meeting FDA standards’ was impossible. Then we started training operators like they were scientists, not just machine pushers. Now we export to 30 countries. It’s not magic. It’s mindset.
Avneet Singh
January 21, 2026 AT 13:53Let’s be real-this entire post is just a glorified compliance brochure. The FDA’s ‘Six Systems’? It’s bureaucratic theater. The real issue is that generic manufacturers are squeezed between price caps and regulatory overreach. No one’s asking how we’re supposed to afford validation studies when the price per tablet is 3 cents. This isn’t about quality-it’s about profit extraction.
Nelly Oruko
January 22, 2026 AT 22:35There’s a quiet truth here: control isn’t enforced-it’s embodied. When every technician knows why their action matters, inspections become irrelevant. The system doesn’t need watching. It’s alive. And that’s rarer than you think.
vishnu priyanka
January 23, 2026 AT 07:36Been there. Saw a facility in Hyderabad get nailed for a 0.5°C deviation in stability storage. They fixed it in 48 hours by installing $200 temperature loggers. FDA came back a month later, smiled, and said ‘Good job.’ No letter. No drama. Just a team that didn’t wait for the audit to care.