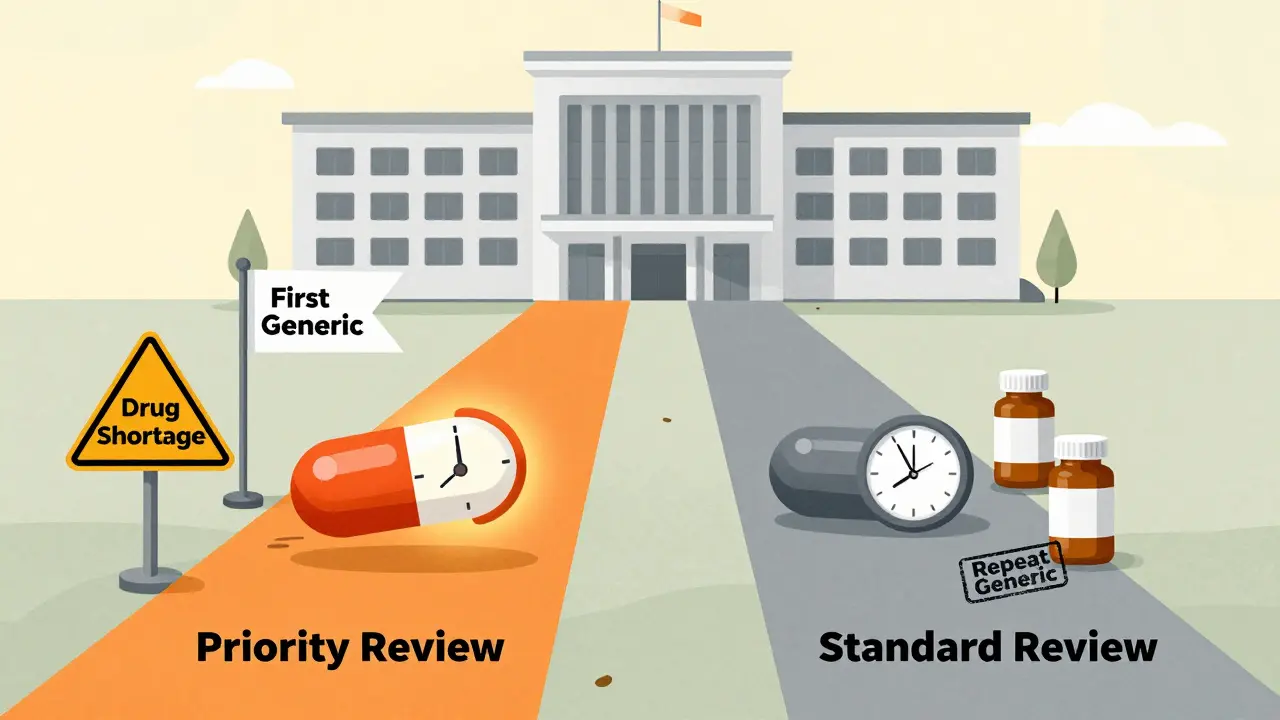
The U.S. Food and Drug Administration (FDA) doesn’t treat every generic drug application the same. Two paths exist: priority review and standard review. The difference isn’t just paperwork-it’s money, timing, and access to life-saving medications. For generic drug makers, getting priority review can mean entering the market two months faster, which often translates to hundreds of millions in extra revenue. For patients, it can mean lower prices and fewer shortages. But how does the FDA decide who gets which path?
What’s the difference between priority and standard review?
The FDA’s review clock starts the moment an Abbreviated New Drug Application (ANDA) is officially filed. Under the current rules, which took effect in October 2022 under GDUFA III, standard review has a target timeline of 10 months. Priority review cuts that to 8 months. That two-month gap might sound small, but in the generic drug world, it’s huge.Standard review applies to the majority of applications-about 70% of them. These are generics for drugs that already have at least one other generic version on the market. Priority review, on the other hand, is reserved for specific cases: first generics (the very first version approved after the brand-name drug’s patent expires), drugs that are in short supply, or complex generics that offer a meaningful improvement over existing options.
It’s not about how big the company is or how much they pay. It’s about impact. A first generic can be the only option for patients for months or even years. If that drug treats a chronic condition like high blood pressure or diabetes, delaying approval by two months means thousands of people pay higher prices longer.
Who qualifies for priority review?
The FDA has clear, public criteria for priority review. The biggest category is first generics. If you’re the first company to submit a complete ANDA after a brand-name drug’s patent or exclusivity expires, you automatically qualify. In 2022, 92.7% of first generics received 180 days of market exclusivity, which is why companies fight hard to be first.The second major category is drugs in shortage. The FDA defines a shortage as a supply that can’t meet patient demand. These aren’t just rare drugs-think insulin, antibiotics, or heart medications. In 2022, nearly 300 drugs were on the FDA’s shortage list. If your generic can fill one of those gaps, you get priority.
There’s also a third, less common path: medically important advances. This applies to complex generics that are harder to make-like inhalers, injectables, or topical creams-and offer better safety, effectiveness, or stability than what’s already available. For example, a generic asthma inhaler that doesn’t use a harmful propellant might qualify.
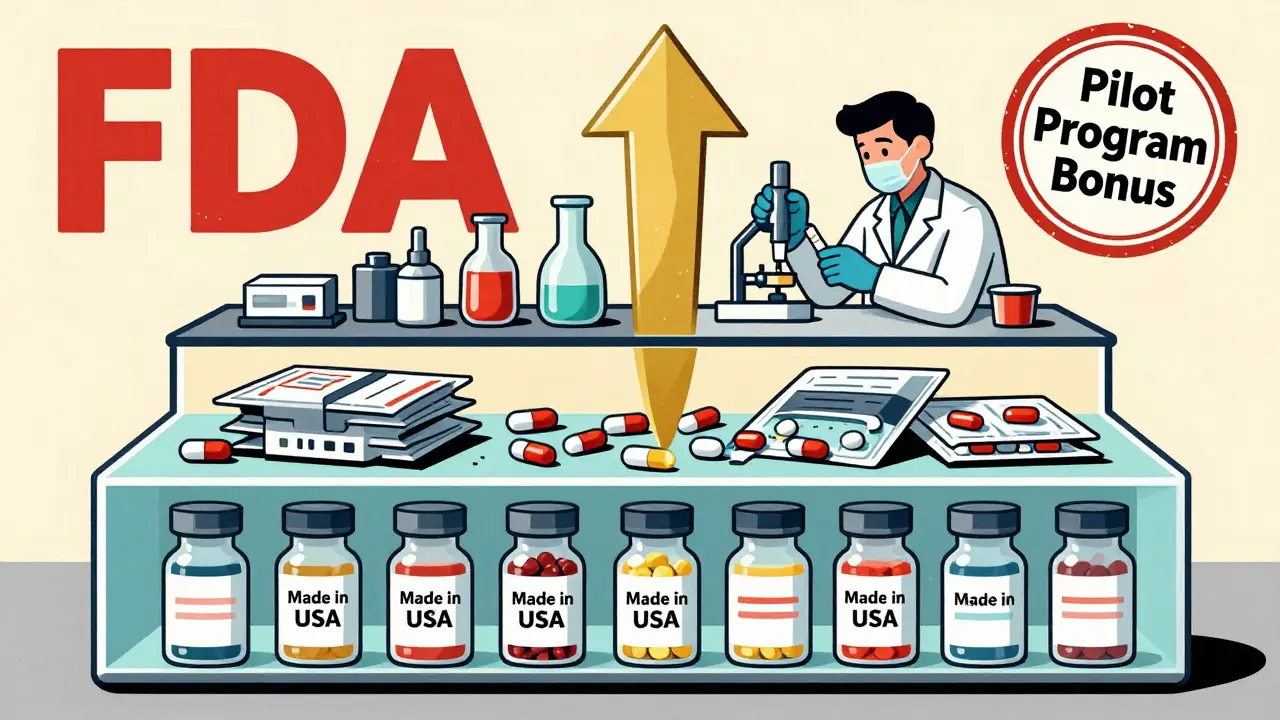
The new U.S. manufacturing bonus
In October 2023, the FDA added a new layer to priority review: the ANDA Prioritization Pilot Program. This isn’t just about speed-it’s about supply chain security.To qualify for this bonus, your generic drug must meet three strict conditions:
- All pivotal bioequivalence testing is done in the U.S.
- The finished dosage form (the pill or injection you swallow) is manufactured in the U.S.
- The active pharmaceutical ingredient (API)-the actual drug substance-is sourced entirely from U.S. suppliers.
This program was created in direct response to the pandemic, when 80% of APIs were made overseas. When global supply chains broke down, U.S. hospitals ran out of critical drugs. The FDA wants to change that.
But here’s the catch: only 12.3% of generic drug companies currently meet all three requirements, according to FDA facility data. Many rely on APIs from India and China, and finished products from Europe. Shifting production to the U.S. is expensive and slow. Still, companies like Teva and Sandoz are already investing millions to upgrade their U.S. labs and factories.
Why does this matter to patients?
The generic drug market is massive. In 2022, generics made up 88.6% of all prescriptions filled in the U.S.-but only 15.3% of total drug spending. That’s how much money patients and insurers save.When a first generic hits the market, brand-name drug prices often drop by 30% to 50% within months. If that drug costs $500 a month, a 40% drop saves $2,400 per patient per year. Multiply that by thousands of users, and you’re talking about hundreds of millions saved.
But delays happen. The average ANDA goes through 1.7 review cycles before approval. Each cycle adds about 4.2 months. Why? Most rejections come from chemistry and manufacturing issues-problems with how the drug is made, not whether it works. That’s why more companies are now meeting with the FDA before submitting their applications. In 2020, only 41% did. By 2023, that number jumped to 63%. The result? First-cycle approval rates rose from 24% to nearly 39%.

What’s next for generic drug approval?
The FDA is testing AI tools to speed up reviews. In internal tests, AI reduced review times for simple applications by nearly 19%. By late 2024, this could become standard for straightforward cases. That means more time for human reviewers to focus on complex applications that need deeper scrutiny.The agency also expects 1,275 ANDA submissions in 2024-up 12.5% from the year before. Priority reviews are projected to make up nearly one-third of all applications. That’s up from 21% in 2020.
The long-term goal? Increase the share of U.S.-made generic drugs from 28% to 40% within five years. That’s not just about speed-it’s about resilience. If the next crisis hits, the U.S. won’t be waiting on shipments from halfway across the world.
What’s the real cost of delay?
A 2-month delay in approval can cost a company $200 million to $500 million in lost revenue, according to regulatory experts. For a small generic maker, that could mean going out of business. For a patient, it might mean paying $100 more a month for a drug they’ve been on for years.And it’s not just about money. The average time from patent expiration to first generic approval is still 2.7 years. That’s too long. Patent litigation, complex manufacturing, and regulatory backlogs all play a role. The FDA’s new programs are trying to cut through that.
The message is clear: the FDA isn’t just reviewing drugs. It’s shaping the future of affordable medicine. Priority review isn’t a favor-it’s a tool to get life-saving drugs to people faster, cheaper, and more reliably.
What is the difference between priority review and standard review for generic drugs?
Priority review for generic drugs takes 8 months from submission, while standard review takes 10 months. Priority review is given to first generics, drugs in shortage, or complex generics that offer a meaningful improvement. Standard review applies to all other generic applications.
How does the FDA decide which generic drugs get priority review?
The FDA grants priority review to applications that are first generics (the first approved after patent expiration), address a drug shortage, or represent a medically important advance over existing versions. The agency checks patent status, shortage lists, and clinical data to determine eligibility.
What is the new U.S. manufacturing pilot program for generic drugs?
Launched in October 2023, the ANDA Prioritization Pilot gives extra review speed to generic drugs that use U.S.-sourced active ingredients, are manufactured in the U.S., and have bioequivalence testing done in the U.S. It’s part of a broader effort to reduce reliance on overseas supply chains.
Why do so many generic drug applications get rejected on first review?
About 32% of ANDAs get a Complete Response Letter (CRL) on first submission. Most rejections are due to chemistry, manufacturing, or controls (CMC) issues-not safety or effectiveness. Problems include inconsistent drug purity, unstable packaging, or flawed bioequivalence data. Pre-submission meetings with the FDA can cut rejection rates in half.
How long does it take for a generic drug to hit the market after patent expiration?
On average, it takes 2.7 years from patent expiration to first generic approval. Delays come from patent lawsuits, complex manufacturing requirements, and FDA review backlogs. Even with priority review, the clock doesn’t start until the application is complete and filed-so preparation matters just as much as speed.
Can a generic drug get priority review if it’s made outside the U.S.?
Yes, but only under the standard priority rules-first generic or drug shortage. The new U.S. manufacturing pilot requires all three components (API, manufacturing, and testing) to be U.S.-based. Without that, companies can still qualify for priority review, but they won’t get the extra boost from the pilot program.
What happens if an ANDA application is incomplete?
If the FDA finds an ANDA incomplete during its 74-day filing review, it issues a Refuse-to-Receive (RTR) letter. The application is not reviewed, and the company must pay a new filing fee-$164,880 in 2024-to resubmit. This resets the clock and can delay approval by months.
How does priority review affect drug prices?
Priority review speeds up market entry, which triggers price competition. First generics typically reduce brand-name drug prices by 30% to 50% within months. Faster approval means patients pay less sooner. In 2022, generics saved the U.S. healthcare system an estimated $378 billion.
Vanessa Barber
January 22, 2026 AT 13:35Everyone acts like priority review is this heroic move, but let’s be real-most of these ‘first generics’ are just copycats with slightly different fillers. The real win is the 180-day exclusivity loophole that lets one company milk the market before anyone else even gets a shot.
Sallie Jane Barnes
January 22, 2026 AT 18:42It is my firm belief that the FDA's prioritization framework represents a commendable alignment of regulatory efficiency with public health imperatives. The structural incentives for domestic manufacturing, in particular, demonstrate a sophisticated understanding of supply chain vulnerability. This is not merely policy-it is public health infrastructure.
Andrew Smirnykh
January 24, 2026 AT 18:16Interesting how the U.S. is pushing for domestic production, but globally, India and China still produce the majority of APIs at a fraction of the cost. I wonder if this policy will just shift the burden to patients through higher drug prices, or if the long-term resilience really balances it out.
charley lopez
January 24, 2026 AT 22:29CMC deficiencies remain the primary bottleneck in ANDA submissions-over 60% of CRLs cite inadequate process validation, impurity profiling, or stability protocol gaps. The pre-submission meeting uptake from 41% to 63% reflects a maturing industry, but the attrition rate at Phase 1 remains structurally high due to under-resourced QA departments in mid-tier firms.
Kerry Evans
January 25, 2026 AT 22:21Of course the FDA is giving priority to U.S.-made drugs-because the rest of the world is just cutting corners. You think India is producing safe meds? Please. They’re using unlicensed labs and skipping dissolution tests. If you want safe drugs, you support American manufacturing-or you’re complicit in endangering patients.
Susannah Green
January 26, 2026 AT 05:09Wait-so if your API is from China, your tablet is made in Germany, and your bioequivalence study was done in Poland… you’re stuck with standard review? That’s wild. I didn’t realize the FDA was this strict about geography. Also, why is the filing fee $164,880?! That’s more than my rent.
Kerry Moore
January 27, 2026 AT 05:20The data regarding the 2.7-year average time from patent expiry to first generic approval is deeply concerning. It suggests that systemic inefficiencies-whether legal, logistical, or regulatory-are undermining the core purpose of the Hatch-Waxman Act. I respectfully urge policymakers to examine the interplay between patent litigation delays and FDA resource allocation.
Sue Stone
January 28, 2026 AT 16:47My grandma’s blood pressure med just got a generic last year. Took forever. Now it’s $12 instead of $200. I’m not mad, I’m just… glad.
Anna Pryde-Smith
January 30, 2026 AT 06:52THE FDA IS A CORRUPT BUREAUCRACY THAT HANDS OUT PRIORITY REVIEWS LIKE CONFECTIONS TO BIG PHARMA! THEY’RE ALL IN ON THE SCAM-THEY LET TEVA AND SANDOZ BUY THEIR WAY IN WHILE SMALL COMPANIES DIE WAITING FOR A SLOTTED REVIEW! THIS ISN’T HEALTHCARE, IT’S A CASINO!